Born With Ebstein’s Anomaly, Jaxon Now Has a Normal Heart
Breann’s phone doesn’t usually ring at 7 in the morning. So when it did—and she saw it was her OB-GYN—she had a bad feeling. She was pregnant, and she had gone for her 20-week ultrasound the day before.
Her doctor did not have good news. “She said something ‘looked off’ on the ultrasound,” Breann remembers. “And it could be nothing—but it could be something.”
Two weeks later, she and her husband, Daniel, were sitting in a pediatric cardiologist’s office, hearing the most devastating news they could imagine. Their unborn baby had Ebstein’s anomaly, a heart defect where the tricuspid valve does not form properly—preventing normal blood flow into the right side of his heart.
“He told us most of these babies don’t even make it to delivery,” Breann says. “And if they do, their lifespan isn’t very long and their quality of life is not good.”
They left the appointment in a daze. “What do we do now?” Daniel remembers thinking. “How the heck do we proceed? Because if there was even a 1% chance for our baby, we wanted to go for it.”
Finding hope
Fortunately, the doctor who had referred them to the cardiologist called later that day and urged them to get a second opinion.
“He told us he could send us anywhere we wanted to go,” Daniel says. “He mentioned Children’s Hospital Los Angeles.”
The couple began doing research and quickly decided on CHLA. It was a long drive but doable—80 miles from their Hesperia home. Most importantly, they liked that the hospital has a Heart Institute that specializes in treating babies and children with the most complex congenital heart defects.
Soon, they were meeting with CHLA Cardiologist Jon Detterich, MD. Dr. Detterich confirmed the Ebstein’s anomaly diagnosis and the risk that the baby would not make it to delivery. But there was also hope.
Their baby would most likely need a surgery called the Starnes procedure, which essentially closes off the malfunctioning right side of the heart. Through a series of three surgeries, a baby’s circulation is rerouted so that the left side of the heart can do all the work—pumping blood to both the body and the lungs.
Children’s Hospital Los Angeles is one of only a few centers in the country to perform this lifesaving procedure, which was developed 30 years ago by Vaughn Starnes, MD, co-Director of CHLA’s Heart Institute.
“The second we met with Dr. Detterich, we knew this was where we wanted to be,” Daniel says. “We were scared, but we felt like we had a plan.”
Now, they just had to get through the rest of the pregnancy.
“It was a roller coaster,” Breann says. “Some days, I’d be poking my belly, going, ‘Are you still alive in there? Why haven’t you kicked? Kick! Kick!’ Because at every appointment, every ultrasound, we didn’t know if he would be gone.”
A severe case
On Feb. 13, 2018, Jaxon arrived—a full-term baby weighing in at 7 pounds, 7 ounces. The first good sign? He was breathing.
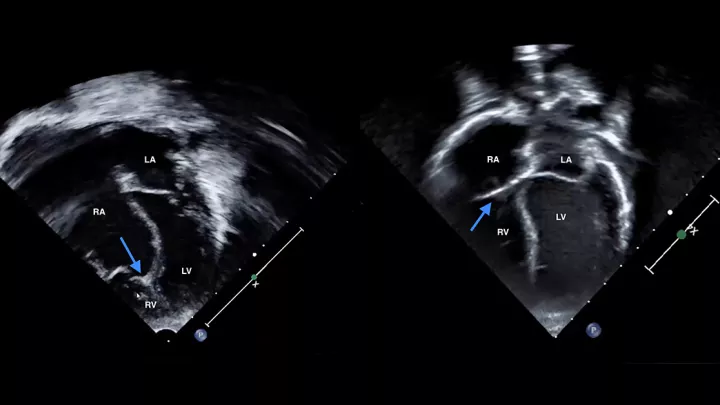
He was quickly transported to Children’s Hospital Los Angeles, where an echocardiogram (ultrasound of the heart) confirmed that his tricuspid valve had not developed normally and was in the wrong position. In addition, his right ventricle—which pumps blood to the lungs—was much smaller than normal, while the right atrium was too large.
There is a spectrum of Ebstein’s anomaly,” Dr. Detterich explains. “Jaxon’s case was severe.”
Doctors immediately started Jaxon on a medicine called prostaglandin E1 (PGE), which keeps open a special fetal blood vessel that normally closes shortly after birth. Keeping this vessel open allowed blood to flow to his lungs despite his abnormal valve.
After about a week, with Jaxon’s lung pressures stable, the team decided to see if Jaxon’s heart could function without the medicine. If it could, he would not need surgery right away. But after just a few hours without PGE, his oxygen levels plummeted—a clear sign that his heart was not able to pump enough blood to his lungs.
Doctors quickly restarted the medicine and began planning for the Starnes procedure. A few days later, on Feb. 26, 2018, Jaxon was wheeled off into the operating room.
Breann—who had suddenly fallen ill the day before—was at home, anxiously glued to her phone. Daniel settled in for a long wait in the hospital, his mom by his side. The surgery was expected to take three hours.
But inside the operating room, things were not going according to plan.
A mid-surgery decision

Dr. Starnes and fellow congenital heart surgeon Ram Subramanyan, MD, PhD, had prepared to close off the right side of Jaxon’s heart. But as Dr. Starnes studied Jaxon’s valve and ventricles, he paused.
“In most of these newborns with severe Ebstein’s anomaly, there’s little to no valve leaflet tissue, and the right ventricle is very thinned out. It’s like a bag without much muscle function to it,” Dr. Starnes explains. “In that situation, there’s not much you can do besides the Starnes procedure.”
But Jaxon’s case wasn’t fitting that mold. Dr. Starnes could see that there was more valve tissue than had been visible on the echocardiograms. That tissue was plastered to the heart wall, as is typical in Ebstein’s anomaly—but it was there.
In addition, the right ventricle wasn’t as weak as it had appeared. “We felt that it could be capable of pumping blood to the lungs if there was a competent tricuspid valve,” Dr. Starnes adds.
The two surgeons conferred. The Starnes procedure has been proven to provide superior outcomes in severe Ebstein’s anomaly. But in this case, Dr. Starnes felt it was possible to repair the valve—giving Jaxon a chance to live with a fully functioning heart.
Dr. Subramanyan agreed. The surgeons decided on the spot to perform an advanced operation called the cone procedure—a surgery rarely done in newborns.
They went to work, carefully separating the valve flaps from the heart wall and rotating them into a cone-shaped valve that could open and close. They also reattached the valve at the correct location in the heart. The procedure took less than an hour.
Dr. Starnes headed out to talk to Daniel, who was startled to see him 90 minutes earlier than expected.
“He came out and said, ‘We fixed it,’” Daniel remembers. “I was like, what do you mean you fixed it? I was so surprised, I don’t even think I said thank you.”
He called Breann. “We were both like, holy cow, what just happened?” Daniel says. “That was a miracle.”
‘A ball of energy’

Jaxon’s parents weren’t the only ones surprised that his valve had been repaired. His cardiologists were astonished, too.
“Most centers do not do this surgery in the newborn period,” Dr. Detterich says. “For our surgeons to make this change on the fly was pretty amazing.”
Jodie Votava-Smith, MD, who led Jaxon’s care for the first week of his life and has been his cardiologist ever since, was also astounded at the news. But a bigger question remained: Would the repair hold up?
The answer was a resounding yes.
“He’s a normal boy,” says his mom. “He’s a ball of energy. You would never know he had surgery.”
Now almost 5, the blonde, tow-headed preschooler loves zipping around on his scooter, swimming, playing with the family dog, having spirited arguments with his 8-year-old sister, Aubree and enthusiastically counting everything around him.
“He’s a numbers guy,” Daniel says with a laugh. “He’ll be like, ‘Dad! There’s five of these!’”
When Jaxon first came home from the hospital, he needed medication for a type of irregular heartbeat that is common in Ebstein’s anomaly. But after a few months, the condition resolved. Today, he takes no medications and has no restrictions.
His yearly echocardiograms at CHLA show a normal heart.
“We thought he was going to have to live with half a heart,” says Dr. Votava-Smith. “Instead, our surgeons fixed it to be a fully functioning, four-chambered heart. It’s a remarkable outcome.”
Breann and Daniel want to express their gratitude to the entire Heart Institute team at CHLA. “I don’t know where else we could have gotten this level of care,” Breann says. “I am beyond thankful to all of his doctors, his nurses, everyone. They are like our family.”