Research Topics
- Retinoblastoma
- Retinal development
- Secondary cancers
- Targeted therapeutics
Research Overview
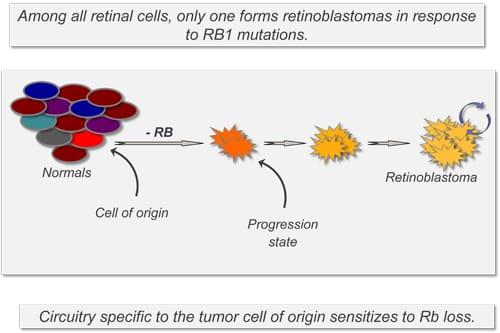
Retinoblastoma is a childhood eye cancer that develops in response to mutations in the RB1 tumor suppressor gene. While RB1 regulates proliferation in many cell types, only cells within the retina routinely form cancers when RB1 function is lost.
Our research aims to characterize the cell type-specific signaling pathways that collaborate with RB1 inactivation, as a means to identify therapeutic targets for retinoblastoma and other cancers.
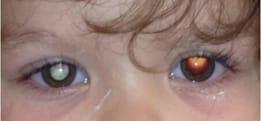
Leukocoria – a white reflection from within the eye – is often the first sign of a growing retinoblastoma tumor.
A Focus on Cone Photoreceptor Circuitry
Our studies focus on the role of cone photoreceptor circuitry in retinoblastoma development. Research performed by others at CHLA in the 1980s showed that retinoblastoma cells express RNAs typical of cone but not rod photoreceptors. More recently, we found that retinoblastoma cells prominently express markers of cones but not other retinal cells, and rely on cone-specific factors for proliferation and survival. We also found that post-mitotic cone precursors have properties that could sensitize to RB1 loss, including unusually high levels of MDM2, N-Myc, and pRB itself, along with dramatically decreased p27, implying that these cells express a pseudo-mitogenic program (Lee et al., 2006, Xu et al., 2009). This year, we showed that Rb is required to suppress retinoblastomagenesis from a cone precursor cell of origin, and that cone-specific circuitry enables the development of Rb-deficient retinoblastoma tumors (Xu et al., 2014).
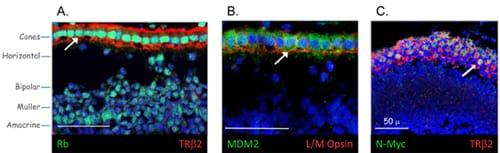
Immunofluorescence images of human retina showing high level expression of Rb (A), MDM2 (B), and N-Myc (C) in post-mitotic cone precursors (marked by TRβ2 or L/M opsin, red). Co-expression of MDM2, N-Myc, and other mitogenic proteins imposes the requirement for Rb’s tumor suppressor function in cone photoreceptor precursors. (Images from Xu et al., 2009, Cell 137, 1018)
Ongoing Studies
Current efforts focus on how cone precursor-related circuitry cooperates with RB1 inactivation to enable the development of retinoblastoma as well as other Rb-deficient tumors. For example, we study how cone factors enable the expression of MDM2 and N-Myc, focusing on a novel MDM2 - N-Myc cross talk applicable to diverse pediatric cancers. We study how a factor (TRβ2) that is especially prominent in cone precursors and pituitary cells collaborates with Rb loss to enable tumors derived from these otherwise unrelated cell types. We study the origin of the human cone precursor mitogenic program, focusing on signaling circuitry specific to human but not mouse cone precursors and critical to retinoblastoma development. Finally, we study how the quasi-differentiated state of post-mitotic cone precursors provides the raw material (in terms of available signaling pathways) to sculpt an oncogenic gene expression program. These studies are expected to identify novel signaling pathways that collaborate with RB1 mutation in diverse human cancers.
Collaborative Efforts
Studies pursued in collaboration with Drs. Jennifer Aparicio, Thomas Lee, and Mark Borchert aim to model retinal development, retinoblastoma, and other retinal developmental disorders in human embryonic stem cell-derived retinas.
Key Findings
- Demonstrated that human cone precursors express especially high levels of Rb together with features of a mitogenic program
- Demonstrated that retinoblastoma cells have properties of human cone precursors, including prominent expression of MDM2 and N-Myc, and rely upon cone-related signaling pathways for their proliferation and survival. Xu et al., 2009.
- Demonstrated that Rb is needed to suppress cone precursor derived retinoblastoma tumors (Xu et al., 2014)
Current Funding
- The Margaret E. Early Medical Research Trust: Molecular Events in Tumor Initiation. This project aims to define cell state transitions and the events that mediate such transitions during the initiation of retinoblastoma tumorigenesis.
- NIH/NCI R01: Cellular Predisposition to Retinoblastoma Tumorigenesis (PI: Cobrinik) This project aims to define cell type-specific circuitry that cooperates with RB1 mutations 1) in the proliferative response to RB1 depletion in human retinal cells, 2) in transgenic mouse retinas with a humanized pattern of MDM2 expression, 3) in retinoblastoma cells, and 4) in the development of mouse pituitary tumors.
- NIH/NCI R43: Development sd-rxRNA as therapy for retinoblastoma and other malignancies (PI: Pavco). This phase 1 SBIR with RXi Pharmaceuticals evaluates the ability of self-delivering RNAi compounds to knockdown gene expression in retinoblastoma tumors, models delivery via intra-ophthalmic artery injection, and defines lead compounds for selected targets.
- RB International: Biomarkers of secondary cancers in retinoblastoma patients (PI: Cobrinik) This project seeks to identify circulating biomarkers of high-frequency secondary cancers in retinoblastoma patients, using patient samples and mouse models.
- In addition to the above awards, we gratefully acknowledge support from the Larry and Cecelia Moh Foundation, the A.B. Reins Foundation, and The Saban Research Institute.