How a Single, Genetic Change Causes Retinal Tumors in Young Children
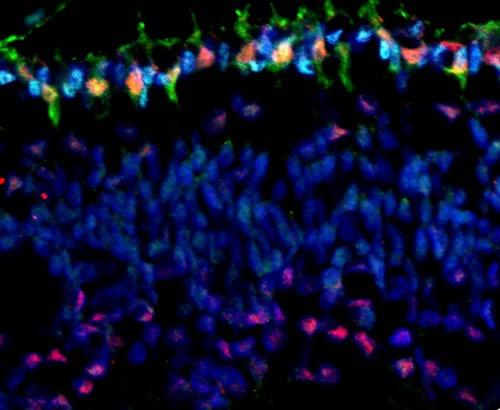
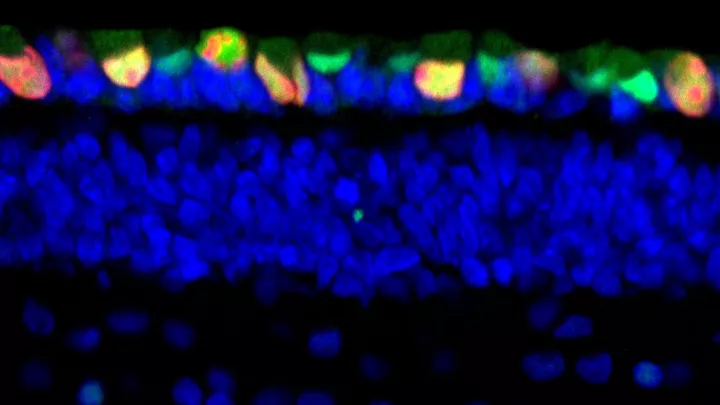
Top image: Cone precursor cells, the photoreceptor cells of the retina that detect color, are in green. The red identifies the Rb protein that is expressed in these cells. This protein is needed to suppress development of retinoblastoma.
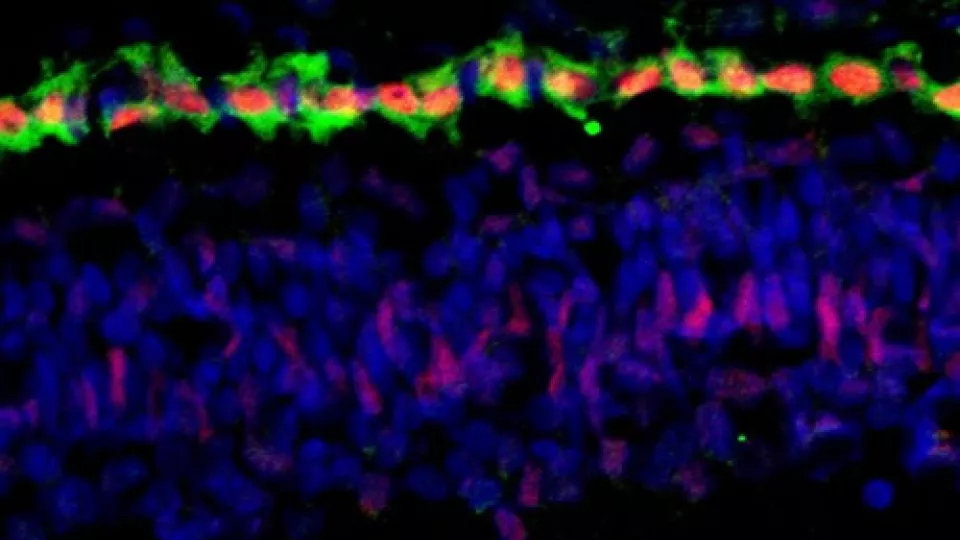
Bottom image: Cones are again in green. However, in this image, the red coloring identifies the MYCN protein that is expressed at especially high levels in cones and promotes development of retinoblastoma when the Rb protein is lost.
______________________________________________________
From their first smile to first day of school, every moment of a child’s young life is captured by their camera-toting parents. But these photographs not only serve as collateral for the troublesome teenage years; they can also be used to identify the most common childhood eye cancer, retinoblastoma. Usually affecting children aged one to two years, this retinal tumor causes the affected eye to glow white in response to a camera’s flash.
While retinoblastoma can be fatal or result in blindness when left untreated, it has also played a special role in understanding the mechanisms of cancer as a whole.
This is because retinoblastoma arises from the mutation of a single gene—the RB1 gene, demonstrating that some cells are only a step away from developing into cancer. (Other cancers, such as skin or breast cancer, arise from an often unpredictable combination of genetic and environmental factors.)
But why do mutations to the RB1 gene only cause tumors of the retina, and not of other cells like those of the iris or pupil?
David E. Cobrinik, MD, PhD, of The Vision Center at Children’s Hospital Los Angeles (CHLA), together with colleagues at Memorial Sloan-Kettering Cancer Center, has recently answered this long-standing question. This study could reveal new cellular signaling pathways that explain retinal and cancer development, and ultimately lead to the development of novel therapies for retinoblastoma. These results were published today in Nature.
In healthy individuals, the RB1 gene codes for a tumor suppressor protein, referred to as Rb, which prevents excessive cell growth. If both alleles of the RB1 gene are mutated early in life, the Rb protein is inactivated. This allows cells to rapidly multiply without resistance and develop into retinoblastoma tumors.
Cobrinik and colleagues discovered that retinoblastomas originate specifically in cone photoreceptor precursors. These are the immature photoreceptor cells in the retina that are responsible for color vision. The researchers also found that the cone precursors prominently express key, cancer-related proteins that enable proliferation and suppress apoptosis, or programmed cell death. This, in combination with the inactivated Rb protein, is sufficient to allow retinoblastomas to form.
“We showed that the cone precursors’ normal developmental program collaborates with RB1 mutations to deregulate cell growth,” said Cobrinik, who is also an associate professor of ophthalmology at the University of Southern California (USC) Eye Institute, Keck School of Medicine of USC. “In other words, loss of the RB1 gene results in abnormal proliferation because the cone precursors lack a self-monitoring ‘surveillance system’ – which would normally cause abnormally proliferating cells to undergo apoptosis. Instead, the cone precursors are able to divide uncontrollably and eventually become cancerous.”