One Step Closer in the Search for an “Autism Gene”
In a condition as heterogeneous as autism spectrum disorder(ASD), finding a shared root of behavioral and neurological symptoms is often a shot in the dark. But in the mid-2000s, researchers found a genetic mutation in one percent of children with ASD, making it the most common cause of this developmental disability.
The Mutation
Called a chr16p11.2 microdeletion, the mutation deletes a large segment of genetic material (30 protein-coding genes and several non-coding RNA genes) in the short arm of chromosome 16. Patients with this deletion show clinical symptoms such as intellectual disability, language impairment, ADHD, and epilepsy, and researchers estimate that 20 to 30 percent of the affected individuals meet the criteria for ASD. Despite its close association with a variety of neurodevelopmental abnormalities, including autism, the direct effects of a chr16p11.2 microdeletion are largely unexplored.
To pinpoint the relationship between this genetic mutation and behavioral symptoms, Di Tian, MD, PhD, neuropathologist in the Department of Pathology and Laboratory Medicine and principal investigator at The Saban Research Institute of Children’s Hospital Los Angeles, examined the first murine model of a chr16p11.2 microdeletion. Results of the study were published earlier this year in Nature Neuroscience.
A Series of Tests
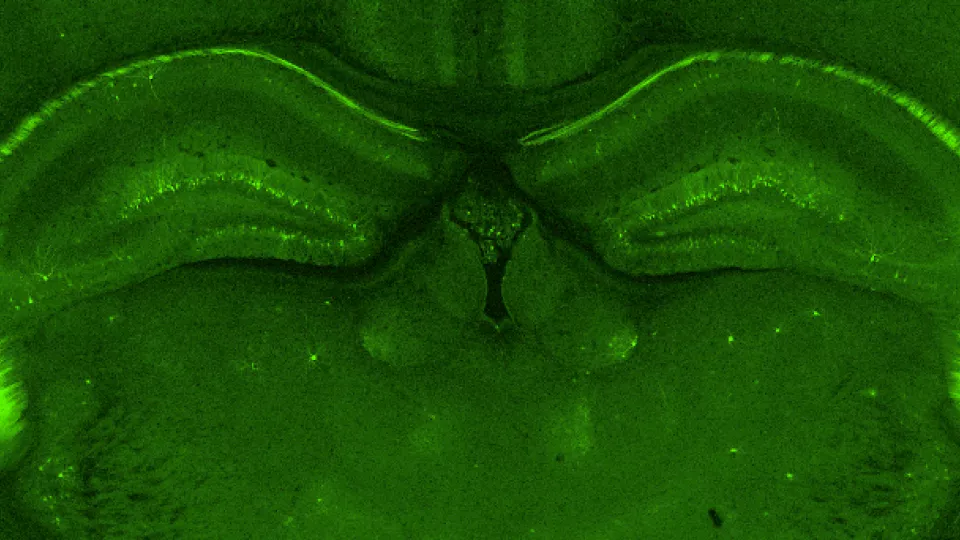
In collaboration with researchers from MIT and Cold Spring Harbor laboratories, Tian focused his attention towards the hippocampus—a brain structure crucial in learning and forming new memories (it’s the area that is most extensively damaged in the brains of people with Alzheimer’s disease). Behavioral tests concluded that 80 to 90 percent of the mutant mice had an impaired ability to learn and remember, and additional tests showed electrophysiological and biochemical abnormalities leading to a decrease in brain plasticity.
“We found that a synaptic protein, Arc, is elevated in the hippocampus of mutant mice, and that this overexpression then causes deficits in a specific type of synaptic plasticity, called mGluR5-medicated long-term depression. Both Arc and mGluR5-mediated synaptic plasticity play important roles in normal brain functions, such as learning and memory, which, as mentioned previously, are significantly impaired in mice with a chr16p11.2 microdeletion,” said Tian.
Reversing the Symptoms
After identifying the problem pathway, Tian and colleagues took their study one step further. They hypothesized that by inhibiting mGluR5 and stopping the overexpression of the Arc protein, they could correct and reverse the behavioral effects in mice with the chr16p11.2 microdeletion—and they were right.
“We found that mice receiving a compound that stops mGluR5, and therefore brings Arc levels down to normal, showed improved learning and memory compared to untreated controls,” said Tian.
While the published results provide an important glimpse into the malfunctioning mechanisms in the brains of those with a chr16p11.2 mutation, additional studies are certainly needed. Tian is currently using a combination of genome-editing techniques, electrophysiology, biochemistry and behavioral testing in an effort to narrow down and identify the exact genes within the deleted region of chr16p11.2 that cause neurodevelopmental symptoms. If successful, this will provide a better understanding of the genetic mechanisms behind autism spectrum disorders and narrow the therapeutic target to precisely reverse the symptoms of ASD.
Image caption: Green fluorescent protein (GFP) stain of the hippocampus highlights the neuronal cell bodies. Image credit: Di Tian, MD, PhD


